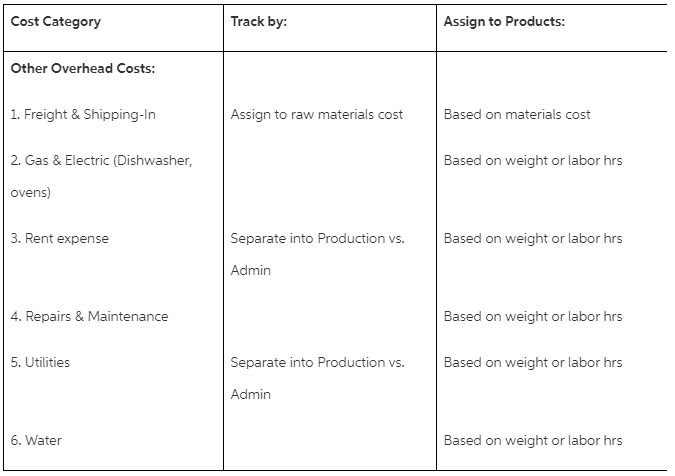
A reactor was charged with THF , 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. Was observed in the overheads indicating complete removal of water from the system.

At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. And sampled for complete formation of imine by HPLC . Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. HPLC results confirmed the reaction was complete by 99.8%.

Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. Ethyl acetate was added to the suspension after it cooled to 20° C. The organic phase was separated and the aqueous phase was checked by HPLC. The organic phase was washed with water , saturated brine solution and concentrated to a small volume under partial vacuum at 45° C. Solid was collected by filtration, rinsed with heptane .

The hydrolysis step was completed by quenching the crude mixture containing primarily intermediate 7 into a premixed solution of 30% aqueous THF containing 6 equiv of triethylamine at ≤0 °C. Prolonged holds of the quenched reaction mixture caused slight degradation and hydrolysis of psilocybin back to psilocin. Typically, this was about 3% conversion after 24 h and up to 15% after a 72 h hold, though high psilocin contamination was found to be tolerated and purged in downstream processing. Following the quench and hold time of at least 60 min, the resulting slurry was filtered, and the filter cake was washed with water to extract remaining 6 adhered to the celite to provide a biphasic filtrate.
The upper organic phase typically contained residual 4 and was removed, with the majority of 6 in the lower aqueous phase. The aqueous phase was transferred back to the reactor, and the crude psilocybin was precipitated by addition of isopropanol followed by distillation of the solvent to a minimum volume. Crude psilocybin solid was subsequently collected by filtration from the aqueous slurry in typically 50–55% yield and 98% purity with psilocin as the highest-level impurity , where most of the psilocin and other process impurities were purged and remained in the filtrate . Oxalyl chloride (8.15 mL) is added dropwise to the cold mixture (10±5° C.) of Thiazole acid 8 (20.18 g) is dissolved in THF and DMF (300 μL) over a period of ˜5 min keeping the internal temperature at 10±5° C.

The cooling bath is removed and the mixture is allowed to reach ambient temperature over a period of ˜30 min. The mixture is stirred at ambient temperature for 30 min to 1 hour. A solution of aniline 7 (19.8 g), DMAP and THF was added at 10±5° C. The ice bath was removed and mixture was heated to 65±2° C.

The mixture was allowed to reach ambient temperature, diluted with EtOAc and washed with water . NaHCO3(5%, 225 mL) was added to the organic portion and the mixture was stirred at ambient temperature for 30 min. The organic portion was concentrated under reduced pressure at approx.

EtOAc was added to the resulting material and the residual water was removed and the mixture was concentrated under reduced pressure at approx. EtOAc was added and the resulting slurry was stirred for 2-6 h and filtered. The solid was washed with EtOAc followed by heptane and air dried for 1 h to give the desired product in 70% yield. Step 4 Oxalyl chloride (8.15 mL) is added dropwise to the cold mixture (10±5 0C) of Thiazole acid 8 (20.18 g) is dissolved in THF and DMF (300 μL) over a period of ~5 min keeping the internal temperature at 10±5°C. The cooling bath is removed and the mixture is allowed to reach ambient temperature over a period of -30 min.

A solution of aniline 7 (19.8 g), DMAP and THF was added at 10±5°C. Et3N (13.2 mL) was added in portions at 10+50C over a period of 10 min. The ice bath was removed and mixture was heated to 65±2°C and stirred overnight . NaHCO3 (5%, 225 mL) was added to the organic portion and the mixture was stirred at ambient temperature for 30 min.

It is a pressure reactor with magnetic drive stirring, inlet & outlet valves, pressure gauge &nitrogen/vent. Solvent & catalyst are charged in the vessel & the slurry formed by mixing istransferred under nitrogen pressure into the reactor. The pressure rating & volume of thissystem is designed depending on the quantity of catalyst, pressure & temperature rating of theautoclave.

Specially designed control system developed by Amar can be offered to charge thecatalyst slurry in continuous mode at a pre-defined flow rate under pressure. This is very usefulfor CSTR, where no suitable pumps are available for slurry. This system is available for anyreactor volume, pressures upto 350 bar & for a wide range materials. Tetrahydrofuran (73 L, 10.7 vol) was charged to the vessel containing 3-(2-chloro-2-oxoacetyl)-1H-indol-4-yl acetate (6.81 kg, 25.6 mol, 1 equiv) under N2 atmosphere and stirred until complete dissolution.

The mixture was cooled to 0–5 °C under a nitrogen atmosphere. A solution of 2 M dimethylamine in THF (13.1 kg, 30.7 mol, 1.2 equiv) was added over 1 h at 0–10 °C via a dip pipe . Triethylamine (3.19 kg, 31.53 mol, 1.23 equiv) diluted in tetrahydrofuran (12.3 L, 1.8 vol) was charged over at least 30 min at 0–10 °C followed by tetrahydrofuran (12.3 L, 1.8 vol) to rinse the addition line.

The mixture was warmed to 15–20 °C and stirred for at least 3 h. Heptane (90.7 L, 13.3 vol) was charged over at least 30 min at 15–20 °C . The mixture was cooled to 0–5 °C over at least 30 min and maintained within this temperature range for at least 1 h. The mixture was filtered under a N2 blanket and washed with heptane at 15–20 °C (27.3 L, 2 vol). The solid was pulled dry under vacuum and nitrogen blanket for at least 15 min until no more filtration liquors were observed. The wet cake was discharged to the vessel and 2-propanol (81.8 L, 12 vol) was added.

The mixture was heated to 80–85 °C and held for 15–60 min until a complete solution was formed. The vessel was cooled down to 18–22 °C over at least 2 h (typically precipitation occurs at 65–70 °C). The slurry was further cooled to 0–5 °C over at least 30 min and maintained within this temperature range for at least 30 min. The mixture was filtered, washed with 2-propanol (13.6 L, 2 vol) at 0–10 °C, and pulled dry under vacuum for at least 15 min.

The solid was washed with water (3 × 17.1 L, 3 × 2.5 vol) at 5–10 °C and then with heptane (2 × 13.6 L, 2 × 2 vol) at 10–20 °C. The solid was dried for at least 16 h in vacuo at 35–40 °C to afford the desired compound (83% yield, 5.83 kg, 99.7% area HPLC). The solid was stored at 20 °C protected from light and moisture. 15.6 g of Type A of Compound , 175 ml of acetone and 3.6 ml of water was added to a 250 ml reactor and heated to 53 degrees C. 900 ul of 10.0 N NaOH was added to reactor and the solution was seeded with Type A. The seeded solution was stirred at 53 degrees C.

A second 900 ul portion of 10.0 N NaOH was added and the system was stirred at 53 degrees C. The final resulting slurry was filtered and the wet solids were washed with 15 ml of acetone. Under vacuum with a nitrogen flow and then exposed the solids to lab air for one hour.

Collected 12.1 g of Compound crystalline sodium salt solids. At room temperature a solution of sodium ethoxide in ethanol (21 weight %; 306 ml) was added to a solution of Compound in THF and water (76.5 ml) while stirring. After stirring for 30 minutes, the mixture was filtered and the filter was washed with THF . The resulting solution was warmed to 65°C and treated with filtered butyl acetate (6640 ml, optionally pre-warmed to 650C) within 30 minutes.

Seeding crystals (0.50 g) were added, and the mixture was stirred at 65°C for 2 hours, while crystallization starts after about 30 minutes. The suspension was cooled to 500C within 1 hour and stirred at this temperature for an additional hour. The title compound was isolated by filtration, washed with filtered butyl acetate (765 ml, optionally pre-warmed to 500C) and dried at 65°C for about 16 h giving Compound crystalline sodium salt (~ 725 g). 15.6 g of Type A of Compound , 175 ml of acetone and 3.6 ml of water was added to a 250 ml reactor and heated to 53 degrees C to dissolve the solids. 900 ul of 10.0 N NaOH was added to reactor and the solution was seeded with Type A. The seeded solution was stirred at 53 degrees C for 10 minutes.

A second 900 ul portion of 10.0 N NaOH was added and the system was stirred at 53 degrees C for 30 minutes over which a slurry developed. The slurry was cooled to 19 degrees C at a cooling rate of 15 degrees C per hour and held overnight at 19 degrees C. Dried solids for 1 hr at 52 degrees C under vacuum with a nitrogen flow and then exposed the solids to lab air for one hour. PS-DVB-derived SILLPs can also be prepared starting from commercial Merrifield resins in the form of beads.

Both gel-type and macroporous resins containing covalently attached imidazolium subunits have shown to act efficiently for the immobilization of CALB, in particular for the polymers having a relatively high content in IL-like fragments. The macroporous resins are, however, more appropriate for being packed into tubular reactors and for the development of flow processes, according to their higher mechanical stability. Again, the nature of the SILLPs regarding both the substitution pattern at the IL-like units and the type of polymeric support used were key factors to obtain an efficient supported biocatalyst.

These elements determine the efficiency of the mass-transfer processes at the interfaces, either of the substrates to the active site or the products from the enzyme. The selection of an IL that contains a large alkyl chain in the cation results in a clear improvement of the efficiency for the biotransformation in monophasic liquid systems and the same takes place when the corresponding SILLPs are prepared. Thus, controlling the structure of the SILLPs, it is possible to develop efficient continuous processes for the transformation of vegetable oil into biodiesel (Fig. 27). At room temperature a solution of sodium ethoxide in ethanol (21 weight %; 306 ml) was added to a solution of Compound in THF and water (76.5 ml) while stiffing.

After stiffing for 30 minutes, the mixture was filtered and the filter was washed with THF . And treated with filtered butyl acetate (6640 ml, optionally pre-warmed to 65° C.) within 30 minutes. Seeding crystals (0.50 g) were added, and the mixture was stirred at 65° C. For 2 hours, while crystallization starts after about 30 minutes. Within 1 hour and stirred at this temperature for an additional hour.

The title compound was isolated by filtration, washed with filtered butyl acetate (765 ml, optionally pre-warmed to 50° C.) and dried at 65° C. For about 16 h giving Compound crystalline sodium salt (˜725 g). Another interesting development of an industrially relevant process is the dimerisation of alkenes, typically propene (Dimersol-G) and butenes (Dimersol-X), to the more valuable branched hexenes and octenes catalysed by Ni complexes. The use of 1,3-dialkylimidazolium chloroaluminates as solvents has been developed and pioneered at IFP. These ILs are good solvents for the butanes and also help in stabilising the Ni catalyst without even requiring an additional ligand.

Under these conditions, the desired products are not soluble in the IL-phase. Thus, it is possible to establish a liquid–liquid biphasic system and the reaction can be performed in a continuous stirred tank-reactor followed by a phase separator. In the separator the upper organic phase containing the products escapes through an overflow for collection and processing, whilst the lower IL-phase containing the catalyst is recycled back to the reactor.

This system dramatically decreases the Ni leaching when compared with the homogeneous Dimersol process in the absence of ILs. The suspension of the mixture of Z and E erythromycin oximes thus obtained is stirred at about 800 rpm for 10 hours or more at a temperature of about 16° C. And 1N HCl solution is added over 1 h 30 min or more so as to bring the pH of the reaction mass to a value of about 9.5. Ethyl acetate is then added and the mass is then heated to about 25-30° C.

After separation of the phases by settling, the aqueous phase is back-extracted with ethyl acetate (2×225 g). The organic phase is rapidly transferred into the reactor and then concentrated under vacuum by partial distillation of the ethyl acetate down to a minimum stirrable volume. The reaction mass obtained is then cooled to about 10° C. The solubility of Compound , as either Type A or the sodium salt form, was investigated in various non-aqueous solvents.

The solutions were prepared by addition of excess Compound to 0.25 ml to 1.0ml of excipient in amber screw cap vials with Teflon lined caps. The samples were allowed to rotate at room temperature for up to 4 days. Sampling was done by centrifuging and filtering through a 0.45 μm PVDF filter. The filtrate was subject to HPLC analysis for determining the solubility.

HPLC analysis was conducted with an Agilent 1100 using gradient or isocratic conditions. Both methods used acetonitrile/water (each with 0.1% Triflouroacetic Acid) and an ACE C- 18 stationary phase with column heating maintained at C. The wavelength of detection was set at 220nm or 264nm. Wet solids were collected and analyzed for form change by XRPD.
The strengths of this technology were highlighted in the previous sections with significant examples that demonstrated the synergies achieved by the combined use of ILs and flow processes. These achievements allow for the development of new greener processes for both the synthesis of high quality ILs and their application in catalytic processes. The IL phases generally contribute to the stabilisation of the catalyst and even, in some cases, to an enhancement of their catalytic activity. Thus, under a continuous flow stream, the productivity can significantly improve. In this way, new opportunities rise from the synergetic combination of these enabling tools.

The greatest opportunities are the design of new processes based on new types of reactors integrating reaction and separation in a single unit. This will contribute to process intensification and it will require the joint effort of chemists and engineers. Regarding the synthesis of ILs, continuous flow synthesis offers a simple, fast and scalable approach for the synthesis of new ILs based on different building blocks. Furthermore, this combination provides exciting opportunities to generate valuable intellectual properties. This isomerisation requires a catalytic Lewis acid and an ionic liquid () as a Lewis base.




